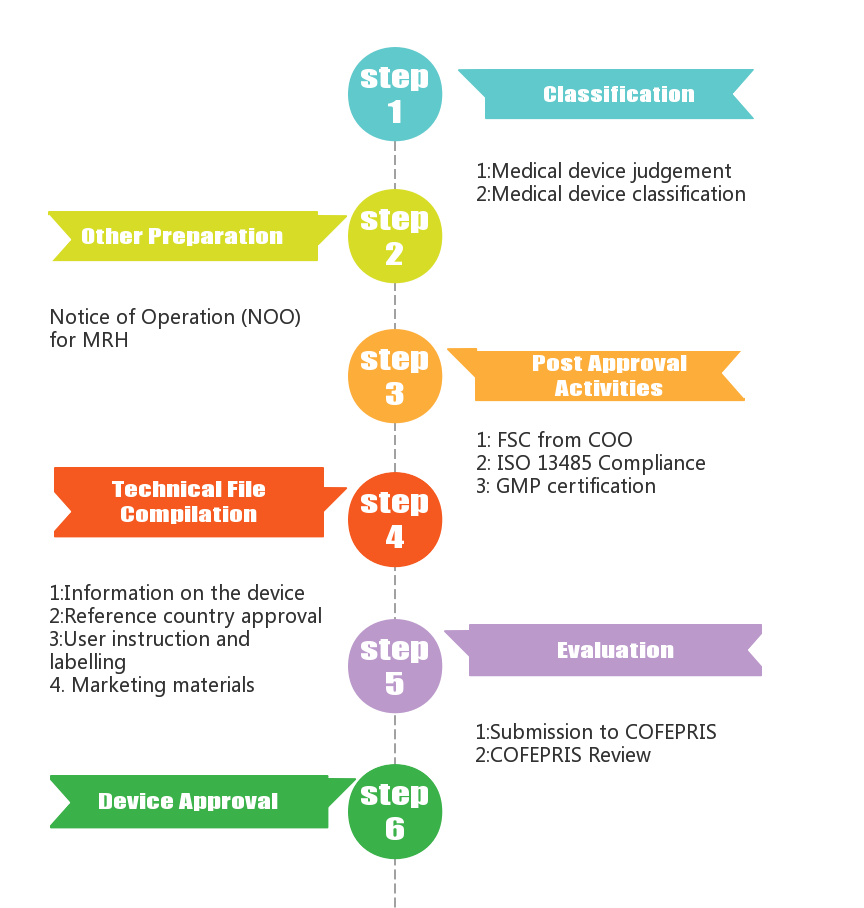
一、主管机构和核心法规
墨西哥的医疗器械受联邦卫生风险保护委员会 (Comisión Federal para la Protección contra Riesgos Sanitarios, COFEPRIS) 监管,主要法规为药事法和医疗器械(Pharmaceutical Affairs Act (PAA) & Regulation for registration of Medical Devices),医疗器械卫生注册有效期为 5 年。
二、产品分类
机构根据其功能和目的将医疗器械分为六大种类:
诊断剂
医疗设备(即配件和电器)
假肢、矫形器和功能性辅助设备
手术材料
卫生设备
牙科用品
根据以上分类,首先判别的产品是否为医疗器械,如果是再进行具体产品风险分类,从而选择正确的认证途径。墨西哥药典 (FEUM) 在医疗器械补编中引入了更新的指南详细说明了产品按风险分类,可分为四类:
Class I低风险
Class I
Class II
Class III