首页
国际项目
美国US
FDA De Novo |
DMF注册 |
NIOSH N95认证 |
UDI |
激光类产品注册 |
化妆品注册 |
食品及接触类产品注册 |
510(K)注册 |
美国代理人 |
FDA注册 |
欧盟EU
CE进口商服务 |
CE-LVD/EMC |
CAPA 体系认证 |
GMPC 体系认证 |
CE-PPE 认证 |
CE-MD 认证 |
欧盟产品注册 |
FSC证书 |
欧盟授权代表 |
CE-IVDR 认证 |
CE-IVDD 认证 |
CE-MDR 认证 |
CE-MDD 认证 |
更多国家
老挝注册 |
缅甸注册 |
埃塞俄比亚注册 |
蒙古注册 |
产品检验检测 |
韩国注册 |
巴西注册 |
澳大利亚注册 |
泰国注册 |
日本注册 |
新加坡注册 |
菲律宾注册 |
马来西亚注册 |
越南注册 |
印度尼西亚注册 |
沙特注册 |
东南亚联盟十国注册 |
哥伦比亚注册 |
加拿大注册 |
中国台湾注册 |
墨西哥注册 |
俄罗斯注册 |
印度注册 |
瑞士注册 |
英国注册 |
体系认证
MDSAP单一审核 |
QSR820 体系认证 |
ISO22716 体系认证 |
ISO15378 体系认证 |
ISO13485 体系认证 |
ISO9001 体系认证 |
中国项目
WHO GDP认证
注册人制度
进口器械注册
国内产品注册
GMP体考
临床及动物试验
EMC摸底测试及整改
新闻资讯
行业动态
常见问题
媒体报道
关于我们
公司简介
发展历程
人才招聘
精英团队
在线认证
联系我们
联系方式
400-0569-812
CN/EN
网站首页
国际项目
美国US
欧盟EU
更多国家
体系认证
中国项目
WHO GDP认证
注册人制度
进口器械注册
国内产品注册
GMP体考
临床及动物试验
EMC摸底测试及整改
新闻资讯
行业动态
常见问题
媒体报道
关于我们
公司简介
发展历程
人才招聘
精英团队
在线认证
联系我们
联系方式
美国US
欧盟EU
更多国家
体系认证
日本的医疗保健市场被誉为东亚最大、最发达的医疗保健系统,对外国医疗器械制造商提出了许多挑战。在人口老龄化、人均收入高以及对高科技产品的巨大需求背景下,以及7% 的预期增长,日本是所有寻求亚洲真正增长的医疗器械制造商需要考虑的市场。
但由于复杂的注册登记流程和语言方面的障碍,日本一直被外国医疗器械制造商认为是挑战性比较高的市场之一。 其实一旦了解清楚其器械注册流程,您会发现它一点儿都不复杂,而且日本作为全世界主要的医疗器械市场,医疗器械市场利润丰厚,回报绝对物有所值。
一、主管机构和核心法规
日本的医疗器械由厚生劳动省(MHLW)的
药品和医疗器械局
(PMDA)根据新修订的药品和医疗器械法 (PMD Act),原药事法(J- PAL全称Pharmaceutical Affairs Law)对医疗器械进行监管。MHLW和PMDA二者的职责如下:
MHLW的主要职责:
证书申请的最终批准
发布指南
监督PMDA活动
PMDA的主要职责:
药品和医疗器械技术评审
GCP、GMP检查
临床试验咨询等
二、产品分类及认证模式
日本根据医疗器械对人体造成可能风险的高低,由低到高,分为四类,即I类、II类、III类和IV类。日本的注册途径取决于各种因素,例如设备类别、分配的 JMDN码、对比设备的可用性以及相关日本工业标准 (JIS) 的可用性。具体注册途径如下所述:
上市前提交(Todokede):
适用于 一般医疗器械(I 类),制造商可以向 PMDA 提交上市前备案。这是一个通知,PMDA 不会进行审查/评估。
上市前认证 (Ninsho):
它适用于 具有相关认证标准 (JIS) 且需要上市前认证的 II 类(和有限数量的 III 类)设备。该过程类似于欧洲 CE 公告机构认证过程,其中医疗器械和质量体系合格评定的审查由 PMDA 外包给第三方认证机构 (RCB)。目前有 14 家 RCB,其中 7 家是主流国际认证公司。申请的平均时间为 3 个月,平均费用为 30,000 美元。
上市前批准(Shonin):
一些没有特定认证标准的 II 类和 III 类设备以及所有 IV 类设备都需要经过上市前批准程序,也称为 Shonin。该申请必须提交给 PMDA 并最终获得 MHLW 的批准。申请处理时间和 PMDA/MHLW 费用从 6 个月和 20,000 美元到 36 个月和 120,000 美元不等,具体取决于分类、JMDN 代码申请和临床证据要求。
三、审核流程
第1步
根据药品和医疗器械法案 (PMD Act) 和日本医疗器械命名法 (JMDN) 代码确定您的器械分类。有5类:
I类 - 一般医疗器械
II 类 - 特定受控医疗器械
II 类 - 受控医疗器械
III 类 - 高度管制的医疗设备*
第IV类 - 高度管制的医疗器械
第2步
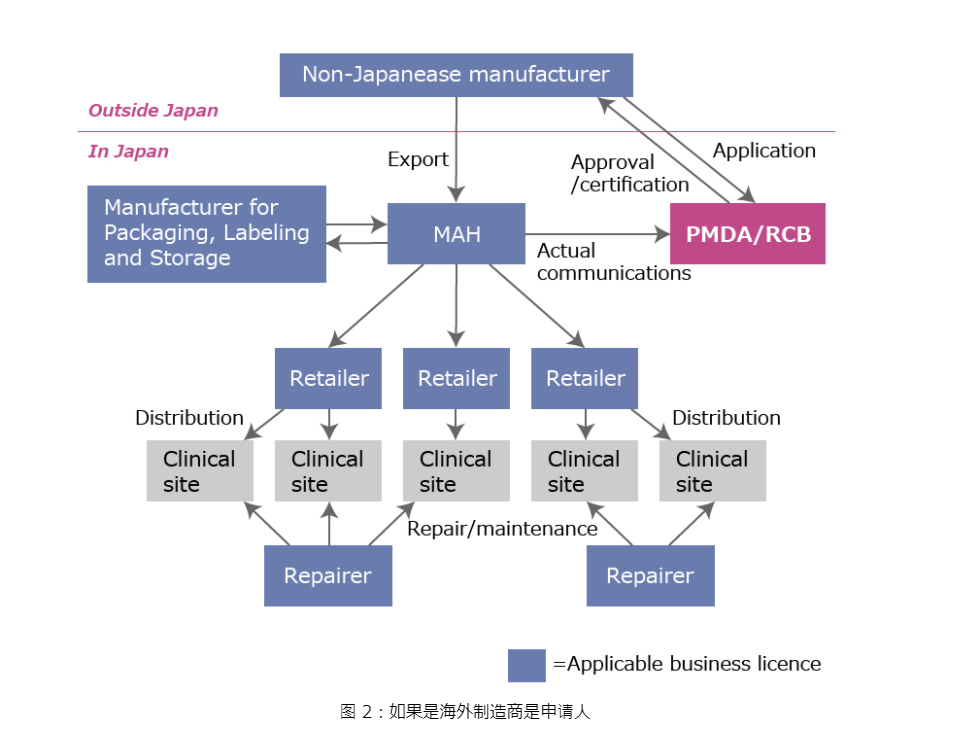
对于 I 类设备,请在日本指定 MAH。
对于所有其他类别,指定营销授权持有人(MAH 或 D-MAH)来管理您在日本的设备注册。您的 MAH 或 D-MAH 将控制您的设备注册。
第3步
所有类别:日本制造商必须向当地县政府注册国内设施。外国制造商必须提交一份
外国制造商注册 (FMR)
向药品和医疗器械管理局 (PMDA) 提出申请。
第4步
所有风险级别产品:实施符合 PMD 法和厚生劳动省 (MHLW) 的质量管理体系 (QMS)法规要求(日本厚生劳动省169法令)。
第5步
对于 I 类设备,提交
上市前提交
制药和医疗器械局 (PMDA)。所有文件必须是日文。
对于 II 类(特定受控)设备,提交
上市前认证
向授权颁发证书的注册认证机构 (RCB) 提出申请。所有文件必须是日文。
对于 II 类(受控)到 IV 类设备,准备
上市前批准
申请以及摘要技术文件 (STED) 格式的注册档案。向 PMDA 提交文件。所有文件必须是日文。
第6步
I 类设备不需要 QMS 审核。
对于 II 类(特定受控)设备,由注册认证机构 (RCB) 进行 QMS 审核。
对于 II 类(受控)到 IV 类设备,PMDA 进行 QMS 审核。对于没有现有 JMDN 代码的“新”设备、IV 类设备和需要临床调查的设备,通常需要进行现场审核。
第7步
对于所有 II 至 IV 类器械,您的 QMS 证书将由 PMDA 或您的注册认证机构颁发。
第8步
对于 II 类(特定受控)设备,RCB 颁发的上市前证书。
对于 II 类(受控)到 IV 类设备,MHLW 颁发的上市前批准证书。
第9步
您现在可以开始在日本营销您的设备。批准不会过期。
注意事项:
市场授权持有人 (MAH)
外国制造商应指定市场授权持有人 (MAH) 作为在日本销售设备的先决条件。但是,PMDA 允许指定指定市场授权持有人 (D-MAH)。在前一种情况下,MAH 拥有并控制产品的注册和证书/批准。在后一种情况下,外国制造商拥有并控制产品的注册和证书/批准,而D-MAH 在日本充当代表。指定D-MAH 而不是 MAH 是理想的,因为更改D-MAH 的过程比更改 MAH 更容易。
外国制造商注册 (FMR)
所有打算将其设备出口到日本的外国制造公司都必须在厚生劳动省 (MHLW) 进行注册。这种注册程序称为外国制造商注册(FMR),以前称为“外国制造商认证(FMA)”或“外国制造商认证(AFM)”。
日本和医疗器械单一审核
计划
(MDSAP)
(医疗器械单一审核计划MDSAP)
允许医疗器械制造商只接受一次审核,即可同时满足多国QMS/GMP要求。日本厚生劳动省(
MHLW
)以及医药医疗器械管理局(
PMDA
)对于产品上市前和上市后的审核,都可使用MDSAP报告,以申请非现场QMS审核或简略非现场QMS审核。MDSAP审核由具备五国监管机构(RA)授权的审核机构(AO)进行。
四、欧必美可提供的服务
推介日本代理MAH/D-MAH服务
协助市场授权持有人监管尽职调查
注册指导/认证咨询(包括生物学、性能测试等各项测试辅导,技术文档编写,临床研究方案(如需)等)
协助市场授权持有人市场授权持有人向 PMDA 或公告机构提交管理
提交前临床试验必要性 (CTN) 咨询
外国制造商注册 (FMR) 指导
数据可靠性检查
质量管理体系检查指导(日本J-QMS/J-GMP, MDSAP 认证支持,包括内部审核、模拟审核、培训等)
标签指导
翻译支持
经销商识别和资格支持
上市后监管方案及实施咨询
注册变更/许可证更新和转让
与市场授权持有人的联络服务
推介仓库制造商或包装制造商完成换标服务
上海欧必美医疗技术集团有限公司 版权所有 Copyright© 2022-2026 All rights reserved
沪ICP备19022935号-1