一、泰国主管机构及法规要求:
泰国是东南亚主要的医疗器械市场之一。泰国的医疗器械受泰国公共卫生部 (MOPH) 下属的泰国食品和药物管理局 (TFDA) 监管。
2021年,泰国食品和药品管理局(TFDA)修订了其医疗器械法规。根据旧法规(2008 年医疗器械法 (BE 2551)) ,90%的器械被归类为通用器械,不需要TFDA详细审查。相反,修订后的法规(医疗器械法/条例 BE 2562 (2019) (第二版)),与东盟地区医疗器械指令(AMDD)一致,大多数器械现在处于TFDA的审查之下,需要通知或注册。
二、产品分类
医疗器械分为四 (4) 类——I、II、III 和 IV。新法规还包括分组规则,允许将医疗设备和 IVD 分组。医疗器械分组是将相关产品组合在一起并在同一申请中注册。
医疗器械分组可分为以下6类:
1. 单品(Single) 2.系统(System) 3. 家庭 (Family)
4.套装 (Set) 5. 诊断试剂盒(IVD Test Kit) 6. 体外诊断产品(IVD Cluster)
三、认证模式
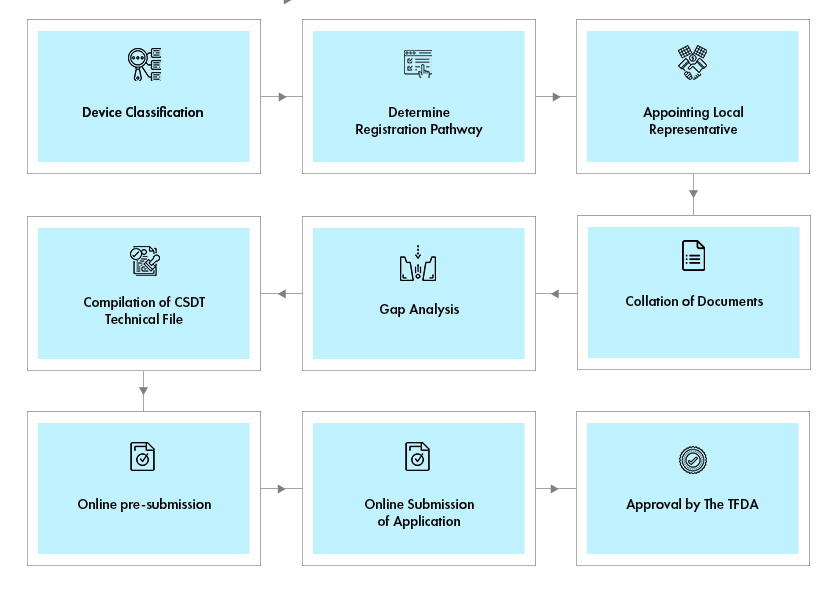
低风险的普通 I 类设备必须在泰国进口和销售之前列名,而 II 类和 III 类设备必须先通过文件预审,IV 类设备必须获得批准的许可证才能投放泰国市场。
II、III 和 IV 类器械需要按照东盟 CSDT 格式提交技术档案。I 类无菌和测量设备需要提交测试报告才能将这些设备投放市场。
1. I类设备备案所需文件:
(1) 载明医疗器械名称和说明、标签、产品规格、医疗器械生产信息或者产品
(2) 所有人详细信息的文件,包括医疗器械附注文件;在有医疗器械文件的情况下。
(3) 在境外注册的境外注册历史文件;
(4) 生产或者进口无菌医疗器械的无菌试验证明文件;
(5) 用于测量的医疗器械生产或者进口时的测试或者校准证明文件;
(6) 产品生产者或者所有者的产品合格证明;
(7) 产品所有人在提交医疗器械进口申请时作为代理人的授权书;
2. II类/III/IV类设备通知所需文件:
(1) 医疗器械的名称和说明文件、医疗器械的标签和标签文件、医疗器械的摘要、医疗器械的生产信息或者产品所有人的详细信息;
(2) 医疗器械安全性和功能性的主要原则及其一致性证明方法的文件;
(3) 风险分析文件;
(4) 质量体系证书;
(5) 产品生产者或者所有者的使用目的、适应症、包装、标签证明和使用方法证明;
(6) 产品生产者或者所有者的产品合格证明;
(7) 生产企业或者产品所有人的医疗器械销售记录证明;
(8) 其他证明文件