首页
国际项目
美国US
FDA De Novo |
DMF注册 |
NIOSH N95认证 |
UDI |
激光类产品注册 |
化妆品注册 |
食品及接触类产品注册 |
510(K)注册 |
美国代理人 |
FDA注册 |
欧盟EU
CE进口商服务 |
CE-LVD/EMC |
CAPA 体系认证 |
GMPC 体系认证 |
CE-PPE 认证 |
CE-MD 认证 |
欧盟产品注册 |
FSC证书 |
欧盟授权代表 |
CE-IVDR 认证 |
CE-IVDD 认证 |
CE-MDR 认证 |
CE-MDD 认证 |
更多国家
老挝注册 |
缅甸注册 |
埃塞俄比亚注册 |
蒙古注册 |
产品检验检测 |
韩国注册 |
巴西注册 |
澳大利亚注册 |
泰国注册 |
日本注册 |
新加坡注册 |
菲律宾注册 |
马来西亚注册 |
越南注册 |
印度尼西亚注册 |
沙特注册 |
东南亚联盟十国注册 |
哥伦比亚注册 |
加拿大注册 |
中国台湾注册 |
墨西哥注册 |
俄罗斯注册 |
印度注册 |
瑞士注册 |
英国注册 |
体系认证
MDSAP单一审核 |
QSR820 体系认证 |
ISO22716 体系认证 |
ISO15378 体系认证 |
ISO13485 体系认证 |
ISO9001 体系认证 |
中国项目
WHO GDP认证
注册人制度
进口器械注册
国内产品注册
GMP体考
临床及动物试验
EMC摸底测试及整改
新闻资讯
行业动态
常见问题
媒体报道
关于我们
公司简介
发展历程
人才招聘
精英团队
在线认证
联系我们
联系方式
400-0569-812
CN/EN
网站首页
国际项目
美国US
欧盟EU
更多国家
体系认证
中国项目
WHO GDP认证
注册人制度
进口器械注册
国内产品注册
GMP体考
临床及动物试验
EMC摸底测试及整改
新闻资讯
行业动态
常见问题
媒体报道
关于我们
公司简介
发展历程
人才招聘
精英团队
在线认证
联系我们
联系方式
美国US
欧盟EU
更多国家
体系认证
想象一下,如果您可以在一个:
拥有 5000 万人口且
被全民医疗保健系统
覆盖的国家;
具有 30 天的国家卫生部 (MoH) 临床试验评估时间表;
对低风险设备进行自动监管市场许可批准,对高风险设备加快 90 天审查;
加入
经合组织
,确保其立法、政策和实践符合全球第一世界标准;
与美国和欧盟签订了自由贸易协定,
并被世界银行营商环境评为
拉丁美洲最容易经商的三大国家之一
,
进行您的早期可行性临床研究并销售您的医疗产品,这是一件多么令人兴奋的事情。
这个国家便是哥伦比亚。
虽然哥伦比亚的经济不比巴西,墨西哥发达,但哥伦比亚医疗器械主要依赖进口,市场需求逐年增长,市场潜力巨大,是海外各医疗器械制造厂商开拓拉美地区市场必争之地。对于众多医疗器械制造商来说,了解哥伦比亚的医疗器械监管规则是获得市场准入,开拓市场的重要条件。
一、主管机构和核心法规
外国制造商在哥伦比亚合法销售其医疗器械之前,他们必须符合哥伦比亚的卫生法规和注册要求。在哥伦比亚,监督医疗器械的监管机构被称为国家药品和食品监督局(INVIMA)。该机构由哥伦比亚卫生部(MoH)设立,不仅包括食品、药品和其他任何卫生产品的监督,而且按2005 年第 4725 号法令还负责对医疗器械的监管。
二、产品分类
在哥伦比亚监管事务终极指南中提到的,INVIMA(国家食品和药品警戒研究所)负责确保在哥伦比亚销售的产品,包括医疗器械、生物医学设备和体外诊断设备,是完整的符合安全和性能要求。
话虽如此,预计每种医疗设备对患者都有固有风险,这取决于一系列因素,包括对身体的侵入程度、预期用途和设备上应用的技术。对于生物医学设备,这种风险级别由其设备分类来区分。
在哥伦比亚,医疗器械可分为以下四类之一:I 类、IIa 类、IIb 类和 III 类,分别代表低风险、中等风险、高风险和极高风险。
根据 2005 年第 4725 号法令第 7 条提供了 18 条分类规则的清单,分 4 章介绍:
无创医疗器械
侵入性医疗器械
适用于有源医疗器械的附加规则
特殊规则
根据 2005 年第 4725 号法令第 6 条,分类基于以下具体标准:
系统的单独设备将被独立评估。
软件将获得与其使用的设备相同的分类。
如果设备打算用于不同的申请,则类别将取决于风险最高的设备。
· 当应用两个或多个分类规则时,必须考虑最高分类。
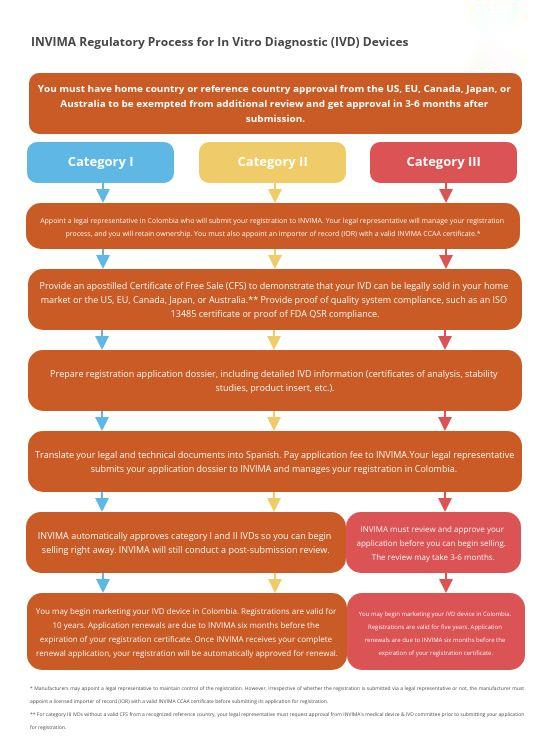
另一方面,IVD也根据技术和各自结果的影响分为三个不同的类别,从高风险的III类开始、中等风险的II类和低风险的I类开始。
2004 年第 3770 号法令的第 4 条列出了 4 条对体外诊断产品进行分类的规则,一般而言,这些设备将所有这些设备分为两大类:与传染性病原体相关的体外诊断设备和与传染性病原体无关的体外诊断设备。
如果任何个人或公司对器械分类犹豫不决,可以咨询欧必美,我们很高兴竭诚为您服务。
三、认证模式
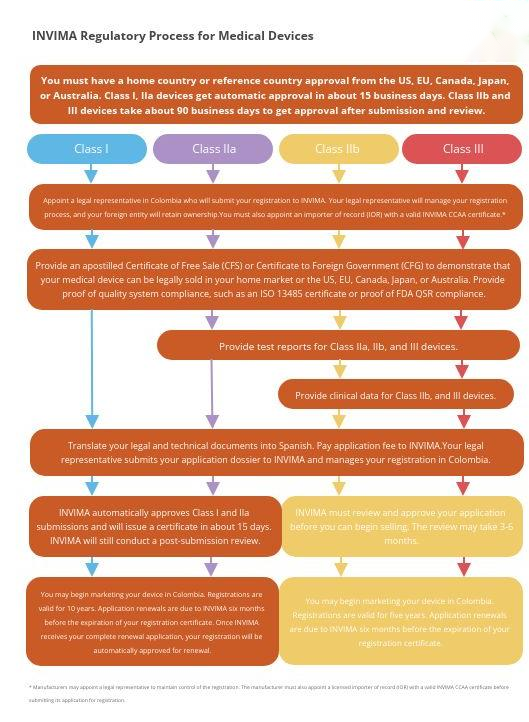
INVIMA将所有医学设备按监管角度分为非受控和受控两类,按不同分类有着各自不同认证模式:
非受控制的 I 类和 lla 类设备有资格进行“自动”注册。受控设备和 IIb 类和 III 类设备需要获得上市前批准。
档案要求
1. 提交 I 类和 lla 类设备的注册文件需要以下文件:
注册申请人的授权委托书
给进口商的授权书
哥伦比亚领事在原产国加盖公章或公证/合法化的自由销售证书
申请表
质量管理体系 (QMS) 证书副本。
医疗器械说明(及其他技术说明)
技术研究和分析测试
最终标签和说明书
如有必要,根据 INVIMA 的要求,提供用于评估医疗器械安全性的附加信息。
2. IIb 类或 III 类非受控设备的注册提交需要以下文件:
除以上文件外,对于生物医疗器械,证据应证明其设计符合适用的标准和规范。
如有必要,根据 INVIMA 的要求,提供用于评估医疗器械安全性的附加信息。
临床研究证明安全性和有效性。
来自原产国或可接受的参考国家(欧盟、美国、加拿大、日本和澳大利亚,或与哥伦比亚有有效相互承认协议的国家)的自由销售证书(CFS)
3. 受控设备
受控设备的申请必须包括以下内容:
1. 符合国际质量标准的证书或证明(制造商和所有合同制造商的 ISO 13485 证书)。
2. 卫生服务贷款机构的名称和地点,IPS,在其中安装设备,或承诺报告设备(如果尚未商业化)
3. 制造商或哥伦比亚代表发布的关于设备的声明
4. 体外诊断产品
最后但并非最不重要的一点是,对于 INVIMA,IVD 以某种方式与医疗设备分开处理,这一点很重要。首先,它们根据风险等级分为三个不同的类别:III 类、II 类和 I 类(分别为高、中和低风险)。
III 类 IVD 确实需要卫生注册才能在哥伦比亚制造、进口、出口、仓储和销售,除非它们在美国、欧盟、加拿大、日本和澳大利亚进行商业化。
II 类和 I 类 IVD 还需要在哥伦比亚制造、进口、出口、仓储和销售时进行自动卫生注册。2004 年第 3770 号法令第 10、12 和 13 条详细规定了申请此注册程序的要求和程序。就像 I 类和 IIa 类的非受控技术生物医学设备一样,自动注册会在提交后几天发布,尽管 INVIMA 将继续评估信息。
周期
医学设备(I 类和 IIa 类)和 I 类和 II 类 IVD 的自动注册:提交信息修改完成后1个月。
医学设备(IIb 类和 III 类注册或商业化许可)以及III 类IVD 注册:收到要求后 4 个月。
对于设备类别 I 和 lla,注册是“自动”的,这意味着将在完成提交后 2-4 天获得。但INVIMA保留自动注册后对提交的信息进行审核的权利;如果 INVIMA 认为需要提交更多信息,则会被要求提交,注册人将有 90 天的时间来遵守。如果不及时提交资料,将导致注册暂停 3 个月,之后根据第 22 条法令 4725/2005取消注册。
有效期
值得一提的是,III类IVD注册有效期为5年。其余的批准,通过医疗器械注册(I类、IIa类、IIb类、III类)、II类和I类IVD以及商业化许可的有效期为10年,这些期限可以延长相应地,通过适当的程序和时间表。
其他体系、测试、标签、临床等具体信息,可致电欧必美或在公众号留意,会有工作人员及时与您联系。
四、审核流程
首先,有必要确定产品是否是:
非可控技术生物医学设备
受控技术生物医学设备或
体外诊断产品
一旦定义了这一点,我们就必须确定分类/类别:
用于医疗器械和生物医学设备:I 类、IIa 类、IIb 类和 III 类
体外诊断设备:I 类、II类和 III 类
医疗器械注册流程
1. 非受控 I 类和 lla
非受控 I 类和 lla 设备的自动批准注册过程将需要以下步骤:
1. 通过授权书任命当地代表。
2. 确保制造商提供的文件符合 INVIMA 要求。
3. 用西班牙语编译技术提交(或至少是西班牙语的测试报告摘要)。
4. 提供产品认证和制造商的测试方法。
5. 向 INVIMA 提交申请。
6. 完成提交后自动获得批准(2-4 天内)。
7. 开始营销。
8. INVIMA 将进行提交后审查。回答 INVIMA 可能就注册提出的任何问题(必须在提出请求后 90 个工作日内提交答复)。
9. 如果 INVIMA 要求,对申请进行任何法律和技术修订。
10. 维持上市后义务。
2. 受控Ib 类和 III 类设备
用于受控器械和 IIb 类和 III 类器械的上市前批准的器械注册过程将需要以下步骤:
1. 通过授权书任命当地代表。
2. 确保制造商提供的文件符合 INVIMA 要求。
3. 用西班牙语编译技术提交。
4. 提供产品认证和制造商的测试方法。
5. 向 INVIMA 提交申请。
6. 回答 INVIMA 可能就注册提出的任何问题(必须在提出请求后 90 个工作日内提交答复)。
7. 批准后,开始营销。
8. 如果 INVIMA 要求,对申请进行任何法律和技术修订。
9. 维持上市后义务。
IVD注册流程
五、欧必美可提供的服务
1.推介哥伦比亚当地经销商
2.电子商务系统帐户创建和管理
3.质量管理体系建立和完善,认证支持,包括内部审核、模拟审核、培训等。
4.器械分类和分组服务
5.与加拿大卫生部的提交前沟通
6.注册指导并代为提交资料,包括准备技术文件,档案汇编 ,辅导测试进行等
7.批准后变更管理指导
8.标签指导服务
9.上市后监管方案及实施咨询
10.许可证更新和转让
上海欧必美医疗技术集团有限公司 版权所有 Copyright© 2022-2026 All rights reserved
沪ICP备19022935号-1